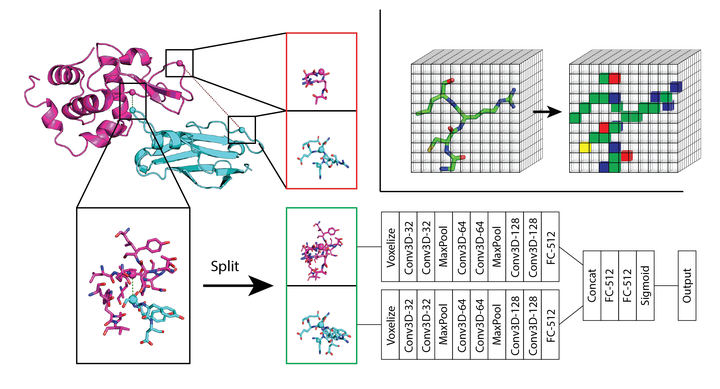
 The SASNet model for interface prediction.
The SASNet model for interface prediction.
Abstract
Despite an explosion in the number of experimentally determined, atomically detailed structures of biomolecules, many critical tasks in structural biology remain data-limited. Whether performance in such tasks can be improved by using large repositories of tangentially related structural data remains an open question. To address this question, we focused on a central problem in biology: predicting how proteins interact with one another—that is, which surfaces of one protein bind to those of another protein. We built a training dataset, the Database of Interacting Protein Structures (DIPS), that contains biases but is two orders of magnitude larger than those used previously. We found that these biases significantly degrade the performance of existing methods on gold-standard data. Hypothesizing that assumptions baked into the hand-crafted features on which these methods depend were the source of the problem, we developed the first end-to-end learning model for protein interface prediction, the Siamese Atomic Surfacelet Network (SASNet). Using only spatial coordinates and identities of atoms, SASNet outperforms state-of-the-art methods trained on gold-standard structural data, even when trained on only 3% of our new dataset.